Development protocol of Digital Sorter
-
ui.R User interface. Use shinydashboard.
-
server.R Where the main codes are.
-
global.R Library all the needed packages.
-
R/ util.R
All the .R files in the R/ folder would be read/sourced automatically by the shiny app. The functions used in the app could be placed in the .R file. Currently, this file includes three functions:
-
get_cell_markers: Get cell surface protein via MSigDB GOCC_CELL_SURFACE set.
Union.cell.surface.marker <- get_cell_markers(category = "C5", subcategory = "GO:CC", set_name = "GOCC_CELL_SURFACE") - ReadRDSFiles: Automatically read all the .rds files in /data folder. (only once when the docker is launched)
- getMarkers: Marker selection function. modified from cerebroApp getMarkerGenes.R.
-
get_cell_markers: Get cell surface protein via MSigDB GOCC_CELL_SURFACE set.
-
www/
All the images in the www/ folder could be sourced by the shiny app with the code
img(src = "file_name.PNG", width=550, height=480). -
data/
All the rds file in the data/ folder would be read by
ReadRDSFilesfunction. Please do not change the names of the following files!!-
Union.cell.surface.marker.rds: a character string of cell surface markers
-
datasets.rds: should be a **SplitObject ** of seurat.
SplitObject(ob, split.by = "dataset_origin") -
markerlist.rds: should be a list including master markers of interested cancer types.
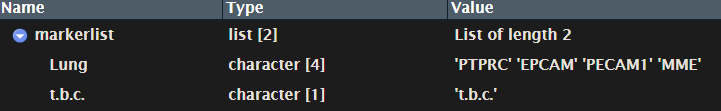
-
The reactive seurat objects in different panels are all independent.
After users select the cohort (dataset), the reactive seurat objects are named ob_reactive_h (for violin plots), ob_reactive1 (for dot plots in gene of interest panel), ob_reactive2 (for dot plots in marker selection panel) in the server.R.
After users select the disease type, the reactive seurat objects are named ob_selected1 (for dot plots in gene of interest panel), ob_selected2 (for dot plots in marker selection panel) in the server.R.