A command-line tool to generate Protein Data Bank (PDB) files with full atomic representation for testing, benchmarking and educational purposes.
![]()
📚 Read the full documentation | Getting Started | API Reference | Tutorials
- Python 3.8+ and basic Python knowledge
- Google Colab account (free) or local Jupyter environment
- Specific tutorials may require domain knowledge (noted in difficulty levels)
| Tutorial | Difficulty | Time | Action |
|---|---|---|---|
| 🤖 AI Protein Data Factory | ⭐ Beginner | 15 min | |
| 🏭 Bulk Dataset Factory | ⭐ Beginner | 15 min | |
| 🔗 Framework Handover | ⭐ Beginner | 10 min | View JAX/PyTorch/MLX Examples |
| ⭕ Macrocycle Design Lab | ⭐⭐ Intermediate | 20 min | |
| 💊 Bio-Active Hormone Lab | ⭐⭐ Intermediate | 20 min | |
| 🔍 Protein Quality Assessment | ⭐⭐ Intermediate | 25 min | |
| 🔬 The Virtual NMR Spectrometer | ⭐⭐ Intermediate | 25 min | |
| 🧲 RDC Alignment Tensor Explorer | ⭐⭐ Intermediate | 30 min | |
| 📡 Neural NMR Pipeline | ⭐⭐ Intermediate | 25 min | |
| 🔗 The NeRF Geometry Lab | ⭐⭐ Intermediate | 25 min | |
| 📏 Geometry Tools Reference | ⭐⭐ Intermediate | 20 min | |
| 🧪 The GFP Molecular Forge | ⭐⭐ Intermediate | 30 min | |
| 🧬 PLM Embeddings (ESM-2) | ⭐⭐ Intermediate | 30 min | |
| 📐 6D Orientogram Lab | ⭐⭐⭐ Advanced | 30 min | |
| 🎯 The Hard Decoy Challenge | ⭐⭐⭐ Advanced | 35 min | |
| 💊 Drug Discovery Pipeline | ⭐⭐⭐ Advanced | 35 min | |
| 🌌 AI Latent Space Explorer | ⭐⭐⭐ Advanced | 35 min | |
| 🏔️ The Live Folding Landscape | ⭐⭐⭐ Advanced | 40 min | |
| ☁️ IDP Conformational Ensembles | ⭐⭐⭐ Advanced | 30 min | |
| 🤖 AlphaFold pLDDT vs NMR S² | ⭐⭐⭐ Advanced | 35 min |
Choose a path based on your background and goals:
Build AI models with synthetic protein data
- 🤖 AI Protein Data Factory (15 min) - Learn zero-copy data handover to PyTorch/JAX
- 🏭 Bulk Dataset Factory (15 min) - Generate thousands of training samples
- 🔗 Framework Handover (10 min) - Integrate with your ML framework
- 🎯 Hard Decoy Challenge (35 min) - Create negative samples for robust training
- 🧬 PLM Embeddings (ESM-2) (30 min) - Add evolutionary context as per-residue node features
- 📐 6D Orientogram Lab (30 min) - Work with rotation-invariant representations
Understand structure, dynamics, and spectroscopy
- 🔗 NeRF Geometry Lab (25 min) - Learn internal coordinate systems
- 📏 Geometry Tools Reference (20 min) - Kabsch, RMSD, and specialized geometry primitives
- 🔬 Virtual NMR Spectrometer (25 min) - Predict relaxation rates and chemical shifts
- 🧲 RDC Alignment Tensor Explorer (30 min) - Visualize the alignment tensor and RDC physics interactively
- 🔍 Protein Quality Assessment (25 min) - Validate structure quality and geometry
- 🧪 GFP Molecular Forge (30 min) - Explore chromophore chemistry
- 🏔️ Live Folding Landscape (40 min) - Visualize energy surfaces and Ramachandran space
- 📡 Neural NMR Pipeline (25 min) - Connect structure to NMR observables
- 🧬 PLM Embeddings (ESM-2) (30 min) - See how sequence encodes secondary structure context
- ☁️ IDP Conformational Ensembles (30 min) - Validate unstructured physical domains
- 🤖 AlphaFold pLDDT vs NMR S² (35 min) - Contrast AI rigidity with physical 15N flexibility
Design and optimize therapeutic peptides
- 💊 Drug Discovery Pipeline (35 min) - End-to-end peptide library to lead selection
- ⭕ Macrocycle Design Lab (20 min) - Create head-to-tail cyclic peptides
- 💊 Bio-Active Hormone Lab (20 min) - Model bioactive peptide hormones
- 🎯 Hard Decoy Challenge (35 min) - Generate decoys for docking validation
- 🌌 AI Latent Space Explorer (35 min) - Navigate chemical space with ML
- 🔬 Virtual NMR Spectrometer (25 min) - Predict experimental observables
- Features
- Installation
- Quick Start
- Usage
- Validation & Refinement
- Output PDB Format
- Scientific Context
- Limitations
- Development
- Glossary of Scientific Terms & Acronyms
- License
✨ Structure Generation
- Full atomic representation with backbone and side-chain heavy atoms + hydrogens
- Customizable sequence (1-letter or 3-letter amino acid codes)
- Random sequence generation with uniform or biologically plausible frequencies
- Conformational diversity: Generate alpha helices, beta sheets, extended chains, or random conformations
- Backbone-Dependent Rotamers: Side-chain conformations are selected based on local secondary structure (Helix/Sheet) to minimize steric clashes (Dunbrack library).
-
Bulk Dataset Generation: Generate thousands of (Structure, Sequence, Contact Map) triplets for AI training via
--mode dataset. - Metal Ion Coordination: Automatic detection and structural injection of cofactors like Zinc (Zn2+) with physics-aware harmonic constraints.
- Disulfide Bonds: Automatic detection and annotation of SSBOND records for Cysteine pairs.
- Salt Bridge Stabilization: Automatic detection of ionic interactions with harmonic restraints in OpenMM.
- Advanced Chemical Shifts: SPARTA-lite prediction + Ring Current Effects (shielding/deshielding from aromatic rings).
-
Relaxation Rates: Lipari-Szabo Model-Free formalism with SASA-modulated Order Parameters (
$S^2$ ), allowing "buried" residues to be more rigid than "exposed" ones. -
Biophysical Realism:
- Backbone-Dependent Rotamers: Chi angles depend on secondary structure.
- Pre-Proline Bias: Residues preceding Proline automatically adopt restricted conformations (extended/beta).
- Cis-Proline Isomerization: X-Pro bonds can adopt cis conformations (~5% probability).
- Post-Translational Modifications: Support for Phosphorylation (SEP, TPO, PTR) with valid physics parameters.
- Cyclic Peptides (Macrocycles): Support for Head-to-Tail cyclization. Closes the peptide bond between N- and C-termini using physics-based minimization.
-
NMR Functionality: As of v1.16.0, all NMR-related features (chemical shifts, relaxation, NOEs, J-couplings) have been refactored into the separate
synth-nmrPython package. This allows for independent use and development of NMR tools.
🚀 High Performance Physics
- Hardware Acceleration: Automatically detects and uses GPU acceleration (CUDA, OpenCL/Metal) if available.
- Apple Silicon Support: Fully supported on M1/M2/M3/M4 chips via OpenCL driver (5x speedup over CPU).
- Vectorized Geometry: Construction kernels are optimized with NumPy vectorization for fast validation.
- Tunable Minimization: Control
toleranceandmax_iterationsto balance speed/quality for bulk datasets.
🔬 Validation Suite
- Bond length validation
- Bond angle validation
- Ramachandran angle checking (phi/psi dihedral angles)
- Side-Chain Rotamer validation (Chi1/Chi2 angles checked against backbone-dependent library)
- Steric clash detection (minimum distance + van der Waals overlap)
- Peptide plane planarity (omega angle)
- Sequence improbability detection (charge clusters, hydrophobic stretches, etc.)
⚙️ Quality Control
--best-of-N: Generate multiple structures and select the one with fewest violations--guarantee-valid: Iteratively generate until a violation-free structure is found--refine-clashes: Iteratively adjust atoms to reduce steric clashes--quality-filter: Use Random Forest-based Structure Quality Filter to validate structure geometry--quality-score-cutoff: Set minimum confidence score for quality filter (0.0-1.0)
📝 Reproducibility
- Command-line parameters stored in PDB header (REMARK 3 records)
- Timestamps in generated filenames and headers
synth-pdb generates structures with realistic properties that mimic real experimental data:
What: Measure atomic mobility/flexibility (columns 61-66)
Formula: B = 8π²⟨u²⟩ (mean square displacement)
Range: 5-60 Ų
Pattern: Backbone (15-25) < Side chains (20-35) < Termini (30-50)
What: Fraction of molecules with atom at position (columns 55-60)
Range: 0.85-1.00
Correlation: High B-factor ↔ Low occupancy
Pattern: Backbone (0.95-1.00) > Side chains (0.85-0.95)
Definition: A Rotamer (Rotational Isomer) is a low-energy, stable conformation of an amino acid side chain defined by specific values of its side-chain dihedral angles (
The "Backbone-Dependent" Twist: The preferred shape of a side chain strongly depends on the shape of the backbone behind it (Alpha Helix vs Beta Sheet).
-
Helix (
$\alpha$ ): Side chains pack tightly. Bulky rotamers (like 'trans' chi1 for Val/Ile) often crash into the backbone (steric clash). -
Sheet (
$\beta$ ): The backbone is extended, creating more room for different rotamers.
Implementation: Synth-PDB uses a simplified version of the Dunbrack Library. It intelligently checks the backbone geometry (
What: Creating a covalent bond between the N-terminal Amine and the C-terminal Carboxyl group to form a closed ring. Biophysical Magnitude:
- Conformational Entropy: Rigidifies the peptide. A linear peptide is a "floppy" string; a cyclic peptide is a "locked" ring. This reduces the entropy loss upon binding to a receptor, significantly increasing affinity.
- Metabolic Stability: Most degradation in the blood happens via exopeptidases (enzymes that clip ends). With no ends to clip, macrocycles are much more stable and long-lived in biological systems.
- Pre-organization: Cyclic peptides are "pre-organized" for their biological function, making them excellent drug scaffolds. Coverage: Supports All 20 Standard Amino Acids (including charged/polar residues).
What: Mirror-images of standard L-amino acids. Biophysical Magnitude:
- Protease Resistance: Most enzymes that degrade proteins (proteases) are "evolutionarily locked" to only recognize L-amino acids. By replacing a single L-amino acid with a D-amino acid, a peptide can become hundreds of times more stable in human blood.
- Bacterial Cell Walls: Bacteria uniquely use D-amino acids (like D-Ala and D-Glu) in their cross-linked peptidoglycan cell walls. This is why many antibiotics (like Penicillin) target these non-L structures.
- Non-Natural Foldamers: D-amino acids allow for the creation of "mirror-image" helices and unique turns (e.g., Beta-turns involving D-Pro) that are impossible with standard biology.
Implementation: synth-pdb mirrors sidechain coordinates across the N-CA-C backbone plane and uses standard PDB 3-letter codes (e.g.,
DAL,DPH).
What: Regular backbone patterns (helices, sheets)
Control: Per-region via --structure parameter
Example: --structure "1-10:alpha,11-15:random,16-25:alpha"
Tip
Realism Equals Efficiency: By using valid backbone angles (Pre-Proline bias) and correct side-chain rotamers, synth-pdb structures start much closer to a physical energy minimum. Validation experiments show this reduces Energy Minimization time by >60% due to fewer initial steric clashes.
What: Realistic backbone geometry validation based on amino acid type using MolProbity/Top8000 data.
- Glycine (GLY): Correctly allowed in left-handed alpha region (phi > 0).
- Proline (PRO): Checks against restricted phi angles.
- General: All other residues are checked against standard Favored/Allowed polygons.
- Precision: Uses point-in-polygon algorithms for accurate classification (Favored, Allowed, Outlier).
What: Natural Extension Reference Frame algorithm
Term: Building 3D structures from "Internal Coordinates" (Z-Matrix)
Mechanism: Places each atom (N, CA, C, O) relative to the local coordinate system of the three previous atoms.
Educational Value: Teaches how math converts 1D sequences + 2D angles into 3D shapes.
What: Structural integration of inorganic ions (e.g. Zinc). Motifs: Detected via ligand clustering (Cys/His sites). Physics: Applied via Harmonic Constraints in Energy Minimization. Importance: Models structural stability of Zinc Fingers and enzymatic sites.
What: Automatic detection of ionic interactions (e.g., LYS+ and ASP-). Criteria: Distance-based detection between charged side-chain atoms (cutoff 5.0 Å). Physics: Stabilized via harmonic restraints during energy minimization. Importance: Maintains tertiary structure integrity in synthetic protein models.
What: Covalent bonds between Cysteine residues Detection: Automatic detection of close CYS-CYS pairs (SG-SG distance 2.0-2.2 Å) Output: SSBOND records added to PDB header Importance: Annotates stabilizing post-translational modifications
What: Binds the N-terminal Nitrogen to the C-terminal Carbon to form a closed ring. Mechanism: Uses OpenMM's physics engine to regularize the covalent bond and minimize ring strain. Bio-Context: Many potent drugs (e.g., Cyclosporine) and toxins are cyclic peptides. Cyclization increases metabolic stability and reduces conformational entropy, improving binding affinity.
synth-pdb is built on the principle of "Code as Textbook".
- Pedagogical Comments: Key source files (
generator.py,test_bfactor.py) contain detailed block comments explaining the why alongside the how (e.g., explaining Lipari-Szabo stiffness vs. B-factor flexibility). - Integrity Safeguards: We include a specialized test suite (
tests/test_docs_integrity.py) that strictly enforces the presence of these educational notes. This ensures that future refactoring never accidentally deletes the scientific context. - Visual Learning: We believe that seeing is understanding. The integrated
--visualizetool connects biophysical theory (minimized energy, restrained dynamics) to immediate visual feedback, helping visual learners grasp complex 3D relationships. - Universal Patterns: The generator is tuned to reproduce universal biophysical phenomena (like terminal fraying and backbone rigidity) rather than just random noise, making it a valid tool for teaching structural biology concepts.
Install the latest stable release from PyPI:
pip install synth-pdbThis installs the synth-pdb package and makes the synth-pdb command available system-wide.
Install directly from the project directory:
git clone https://github.com/elkins/synth-pdb.git
cd synth-pdb
pip install .- Python 3.8+
- NumPy
- Biotite (for residue templates and structure manipulation)
Dependencies are automatically installed with pip.
Generate a simple 10-residue peptide:
synth-pdb --length 10Generate and validate a specific sequence:
synth-pdb --sequence "ACDEFGHIKLMNPQRSTVWY" --validate --output my_peptide.pdbGenerate with mixed secondary structures and visualize:
synth-pdb --structure "1-10:alpha,11-20:beta" --visualizeGenerate the best of 10 attempts with clash refinement:
synth-pdb --length 20 --best-of-N 10 --refine-clashes 5 --output refined_peptide.pdbGenerating "good" structures is only half the battle. To train robust AI models (like AlphaFold-3 or RosettaFold), researchers need High-Quality Negative Samples—structures that look physically plausible but are biologically or topologically incorrect.
Synth-PDB provides three powerful mechanisms for generating these "Hard Decoys":
Force a specific sequence onto the backbone "fold" of a completely different sequence. This creates a realistic-looking structure where the side-chain packing is fundamentally incompatible with the backbone.
# Thread Poly-Ala sequence onto a backbone generated for Poly-Pro
synth-pdb --mode decoys --sequence AAAAA --template-sequence PPPPP --hardAdd controlled, random noise to ideal Ramachandran angles. This creates "near-native" decoys—structures that are almost correct but have subtle, realistic errors.
# Add 5 degrees of maximum drift to all phi/psi angles
synth-pdb --mode decoys --drift 5.0Generate a perfectly valid structure for a sequence, then randomly shuffle the identity of the residues in the final PDB. This tests if an AI model can detect that a residue (e.g., Trp) is in an environment meant for another (e.g., Gly).
synth-pdb --mode decoys --sequence ACDEF --hard --shuffle-sequenceMost synthetic PDB generators create static bricks. They might create reasonable geometry, but the "B-factor" column (Column 11) is often just zero or random noise.
Synth-PDB is different. It simulates the physics of protein motion to generate a unified model of structure AND dynamics.
We implement the Lipari-Szabo Model-Free formalism (Nobel-adjacent physics) directly into the generator:
-
Structure Awareness: The engine analyzes the generated geometry (
alpha-helixvsrandom-coil). -
Order Parameter (
$S^2$ ) Prediction: It assigns specific rigidity values:-
Helices:
$S^2 \approx 0.85$ (Rigid H-bond network) -
Loops:
$S^2 \approx 0.65$ (Flexible nanosecond motions) -
Termini:
$S^2 \approx 0.45$ (Disordered fraying)
-
Helices:
-
Unified Output:
-
PDB B-Factors: Calculated via
$B \propto (1 - S^2)$ . When you visualize the PDB in PyMOL, flexible regions visually appear thicker/redder, matching real crystal data distributions. -
NMR Relaxation:
$R_1, R_2, NOE$ rates are calculated from the same parameters.
-
PDB B-Factors: Calculated via
Why this matters:
"The correlation between NMR order parameters (
$S^2$ ) and crystallographic B-factors is a bridge between solution-state and solid-state dynamics." — Fenwick et al., PNAS (2014)
This feature allows you to test bioinformatics pipelines that rely on correlation between sequence, structure, and experimental observables, without needing expensive Molecular Dynamics (MD) simulations.
Generate relaxation rates (
python main.py relax --input output/my_peptide.pdb --output output/relaxation_data.nef --field 600 --tm 10.0This module now implements the Lipari-Szabo Model-Free formalism with structure-based Order Parameter (
-
Helices/Sheets:
$S^2 \approx 0.85$ (Rigid, high$R_1/R_2$ ) -
Loops/Turns:
$S^2 \approx 0.65$ (Flexible, lower$R_1/R_2$ ) -
Termini:
$S^2 \approx 0.45$ (Highly disordered)
This creates realistic "relaxation gradients" along the sequence, perfect for testing dynamics software.
Want to see the Physics + Visualization capabilities in action?
Run this command to generate a Leucine Zipper (classic alpha helix), minimize its energy using OpenMM, and immediately visualize it in your browser:
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" --conformation alpha --minimize --visualizeThis effectively demonstrates:
- Generation: Creating the alpha-helical backbone.
- Minimization: "Relaxing" the structure (geometry regularization).
- Visualization: Launching the interactive 3D viewer.
-
--length <LENGTH>: Number of residues in the peptide chain- Type: Integer
- Default:
10 - Example:
--length 50
-
--sequence <SEQUENCE>: Specify an exact amino acid sequence- Formats:
- 1-letter codes:
"ACDEFG" - 3-letter codes:
"ALA-CYS-ASP-GLU-PHE-GLY"
- 1-letter codes:
- Overrides
--length - Example:
--sequence "MVHLTPEEK"
- Formats:
-
--plausible-frequencies: Use biologically realistic amino acid frequencies for random generation- Based on natural protein composition
- Ignored if
--sequenceis provided
-
--conformation \u003cCONFORMATION\u003e: Secondary structure conformation to generate- Options:
alpha,beta,ppii,extended,random - Default:
alpha(alpha helix) - Choices:
alpha: Alpha helix (φ=-57°, ψ=-47°)beta: Beta sheet (φ=-135°, ψ=135°)ppii: Polyproline II helix (φ=-75°, ψ=145°)extended: Extended/stretched conformation (φ=-120°, ψ=120°)random: Random sampling from allowed Ramachandran regions
- Example:
--conformation beta
- Options:
synth-pdb serves as valid data generator for training Deep Learning models (GNNs, Transformers, Diffusion Models). It can generate massive, diverse, and labeled datasets.
Command:
synth-pdb --mode dataset --dataset-format npz --num-samples 1000 --output my_training_dataFeatures:
- Formats:
npz: (Recommended) Compressed NumPy archives. Containscoords(L,5,3),sequence(One-hot), andcontact_map(LxL). Ideal for PyTorch/TensorFlow dataloaders.pdb: Writes individual PDB files and CASP contact maps (slower, for legacy tools).
- Multiprocessing: Automatically uses all available CPU cores.
- Manifest: Generates a
dataset_manifest.csvtracking all samples and their metadata (split, length, conformation).
Output Structure (--dataset-format npz):
my_training_data/
├── dataset_manifest.csv
├── train/
│ ├── synth_000001.npz
│ ├── synth_000002.npz
│ ...
└── test/
├── synth_000801.npz
...
-
--validate: Run validation checks on the generated structure- Checks: bond lengths, bond angles, Ramachandran, steric clashes, peptide planes, sequence improbabilities
- Reports violations to console
-
--guarantee-valid: Generate structures until one with zero violations is found- Implies
--validate - Use with
--max-attemptsto limit iterations - Example:
--guarantee-valid --max-attempts 100
- Implies
-
--max-attempts <N>: Maximum generation attempts for--guarantee-valid- Default:
100
- Default:
-
--best-of-N <N>: Generate N structures and select the one with fewest violations- Implies
--validate - Overrides
--guarantee-valid - Example:
--best-of-N 20
- Implies
-
--refine-clashes <ITERATIONS>: Iteratively adjust atoms to reduce steric clashes- Applies after structure selection
- Iterates until improvements stop or max iterations reached
- Example:
--refine-clashes 10
Note
Despite the flag name history, this feature uses a classical Random Forest classifier (scikit-learn), not a neural network or generative AI. It scores structures on geometric quality metrics derived from Ramachandran angles, steric clashes, bond lengths, and radius of gyration.
-
--quality-filter: Enable the Structure Quality Filter to screen generated structures.- Using a Random Forest classifier trained on thousands of samples, this filter automatically rejects "low quality" structures (clashing, distorted geometry).
- It considers Ramachandran angles, steric clashes, bond lengths, and radius of gyration.
- Useful for filtering out failed minimization attempts in bulk generation.
-
--quality-score-cutoff <FLOAT>: Minimum probability score (0.0-1.0) for a structure to be considered "Good".- Higher values = stricter filtering (fewer false positives, more false negatives).
- Default:
0.5 - Example:
--quality-score-cutoff 0.8(Only keep highly confident good structures) - Scores below
0.5are typically rejected as "Bad".
-
--minimize: Run physics-based energy minimization (OpenMM).- Defaults to implicit solvent (OBC2) and AMBER forcefield.
- Highly recommended for "realistic" geometry.
- Example:
--minimize
-
--solvent <MODEL>: Specify the solvent model for minimization/equilibration.- Options:
obc2(default),obc1,gbn,gbn2,hct,explicit - Example:
--solvent explicit(simulates a TIP3P water box)
- Options:
-
--solvent-padding <FLOAT>: Padding distance (in nm) for the explicit water box.- Default:
1.0 - Example:
--solvent-padding 1.5
- Default:
-
--keep-solvent: Retain the generated water molecules (HOH) in the final PDB file.- Default: False (water is stripped for cleaner outputs)
-
--optimize: Run Monte Carlo side-chain optimization.- Reduces steric clashes by rotating side chains.
- Example:
--optimize
-
--forcefield <NAME>: Specify OpenMM forcefield.- Default:
amber14-all.xml - Example:
--forcefield amber14-all.xml - Default:
amber14-all.xml
- Default:
-
--minimization-k <FLOAT>: Energy minimization tolerance (kJ/mole/nm).- Higher values = Faster but less precise.
- Recommended for bulk generation:
100.0 - Default:
10.0(High Precision)
-
--minimization-max-iter <INT>: Max iterations for minimization.0= Unlimited (Convergence based on tolerance)- Recommended for bulk generation:
1000 - Default:
0
📦 NMR Functionality Powered by
synth-nmr
As of version 1.17.0, all NMR-related functionality (NOE calculation, relaxation rates, chemical shifts, J-couplings) is provided by the standalonesynth-nmrpackage. This package can be used independently for NMR data generation in your own projects. The integration is fully backward compatible—all existing code continues to work without changes.
-
--gen-nef: Generate synthetic NOE restraints in NEF format.- Scans structure for H-H pairs < cutoff.
- Outputs
.neffile. - Note: Requires hydrogens (use with
--minimizeor internal default).
-
--noe-cutoff <DIST>: Cutoff distance for NOEs in Angstroms.- Default:
5.0 - Example:
--noe-cutoff 6.0
- Default:
-
--nef-output <FILE>: Custom output filename for NEF.
-
--gen-relax: Generate synthetic NMR relaxation data ($R_1, R_2, {^1H}-^{15}N\ NOE$ ) in NEF format.- Calculates Model-Free parameters (
$S^2 \approx 0.85$ for core,$0.5$ for flexible termini). - Outputs
_relax.neffile. -
Physics Note:
$NOE$ values depend on tumbling time, not just internal flexibility.
- Calculates Model-Free parameters (
-
--field <MHZ>: Proton Larmor frequency in MHz.- Default:
600.0 - Calculates proper spectral density frequencies for this field.
- Default:
-
--tumbling-time <NS>: Global rotational correlation time ($\tau_m$ ) in nanoseconds.- Default:
10.0 - Controls the overall magnitude of relaxation rates. Larger proteins have larger
$\tau_m$ .
- Default:
-
--export-constraints <FILE>: Export contact map constraints for modeling/folding.- Useful for checking agreement with AlphaFold/CASP predictions.
- Outputs a file containing residue-residue contacts.
- Example:
--export-constraints constraints.casp
-
--constraint-format {casp,csv}: Format for the exported constraints.casp: Critical Assessment of Structure Prediction (RR) format.csv: Comma-separated values (i, j, distance).- Default:
casp
-
--constraint-cutoff <DIST>: Distance cutoff for defining binary contacts (Angstroms).- Default:
8.0
- Default:
-
--export-torsion <FILE>: Export backbone torsion angles (Phi, Psi, Omega) for every residue.- Useful for training ML models on backbone geometry.
- Outputs a CSV or JSON file.
- Example:
--export-torsion angles.csv
-
--torsion-format {csv,json}: Format for the exported data.- Default:
csv
- Default:
-
--gen-msa: Generate a Multiple Sequence Alignment (MSA) by simulating neutral drift.- Conserves hydrophobic core residues while mutating surface residues.
- Outputs a FASTA file useful for testing co-evolution signals in AI models.
-
--msa-depth <N>: Number of sequences to generate.- Default:
100
- Default:
-
--mutation-rate <RATE>: Probability of mutation per position per sequence.- Default:
0.1(10% divergence per sequence).
- Default:
-
--export-distogram <FILE>: Export NxN Distance Matrix representing the protein geometry.- Rotation-invariant representation ideal for AI model training/validation.
- Supports
json,csv, ornpz(NumPy) formats. - Example:
--export-distogram dist.json
-
--distogram-format {json,csv,npz}: Output format.- Default:
json
- Default:
-
--ph <VAL>: Set pH for titration (default 7.4).- Automatically adjusts Histidine protonation (
HIS$\rightarrow$ HIPif pH < 6.0). - Critical for realistic electrostatics and NMR chemical shifts.
- Automatically adjusts Histidine protonation (
-
--cap-termini: Add terminal blocking groups.- N-terminus: Acetyl (
ACE) - C-terminus: N-methylamide (
NME) - Removes charged termini (
$\text{NH}_3^+$ /$\text{COO}^-$) for realistic peptide modeling.
- N-terminus: Acetyl (
-
--cyclic: Generate a Head-to-Tail cyclic peptide.- Connects the N-terminus and C-terminus with a covalent peptide bond.
-
Requirement: Automatically implies
--minimizeto ensure proper closure. -
Incompatibility: Disables
--cap-termini.
-
--equilibrate: Run Molecular Dynamics (MD) equilibration.- Simulates the protein at 300 Kelvin (solution state).
- Uses Langevin Dynamics to shake atoms out of local minima.
- Generates a "thermalized" structure closer to NMR conditions.
- Options:
--md-steps <INT>(default 1000,$\approx$ 2 ps).
-
--metal-ions {auto,none}: Control metal ion coordination.-
auto(default): Scans for binding sites and injects ions. -
none: Disables automatic coordination.
-
-
--phosphorylation-rate <FLOAT>: Probability of phosphorylating S/T/Y residues.- Value between 0.0 and 1.0.
- Converts SER->SEP, THR->TPO, TYR->PTR.
- Mimics kinase activity for regulatory simulation.
- Example:
--phosphorylation-rate 0.5
-
--cis-proline-frequency <FLOAT>: Probability of X-Pro peptide bond being Cis.- Default:
0.05(5%) - Cis-Proline is critical for tight turns and folding.
- Set to
0.0for all-Trans,1.0for all-Cis.
- Default:
--mode dataset: Enable bulk generation mode.--num-samples <N>: Number of samples to generate (default 100).--min-length <N>,--max-length <N>: Range for random sequence lengths (default 10-50).--train-ratio <FLOAT>: Fraction of samples for the training set (default 0.8).--output <DIR>: Directory to save the dataset.
-
--output <FILENAME>: Custom output filename- If omitted, auto-generates:
random_linear_peptide_<length>_<timestamp>.pdb - Example:
--output my_protein.pdb
- If omitted, auto-generates:
-
--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}: Logging verbosity- Default:
INFO - Use
DEBUGfor detailed validation reports
- Default:
-
--seed <INT>: Random seed for reproducible generation- Default:
None(Random) - Example:
--seed 42 - Guarantees identical output for the same command.
- Default:
-
--help: Show the help message and exit.
# Simple 25-residue peptide
synth-pdb --length 25
# Custom sequence with validation
synth-pdb --sequence "ELVIS" --validate --output elvis.pdb
# Use biologically realistic frequencies
synth-pdb --length 100 --plausible-frequencies
# Generate a random 20-residue alpha helix
synth_pdb --length 20 --conformation alpha --output random_helix.pdb
# Generate a high-quality, physically realistic structure (Recommended)
# Includes: Minimization, Terminal Capping, and Thermal Equilibration (MD)
synth_pdb --length 20 --minimize --cap-termini --equilibrate --output best_structure.pdb
# Generate beta sheet conformation
synth-pdb --length 20 --conformation beta --output beta_sheet.pdb
# Generate extended conformation
synth-pdb --length 15 --conformation extended
# Generate random conformation (mixed alpha/beta regions)
synth-pdb --length 30 --conformation random
# 🤖 Bulk dataset generation for AI training
synth-pdb --mode dataset --num-samples 500 --min-length 10 --max-length 40 --output ./my_dataset
# ⛓️ Generate a Zinc Finger with structural cofactors
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --minimize --metal-ions auto --output zinc_finger.pdb# Generate until valid (may take time!)
synth-pdb --length 15 --guarantee-valid --max-attempts 200 --output valid.pdb
# Best of 50 attempts
synth-pdb --length 20 --best-of-N 50 --output best_structure.pdbSimulate your protein in a realistic water box (TIP3P) for high-fidelity physics or export the explicit solvent map for downstream molecular dynamics.
# Basic explicit solvent: generate a small peptide and pad with 1.2 nm of water.
# By default, synth-pdb strips the water atoms before saving the final clean PDB.
synth-pdb --sequence ALA-PRO-GLY --minimize --solvent explicit --solvent-padding 1.2 --output small_peptide.pdb
# Retain the water box: save the entire simulated system (protein + thousands of HOH atoms)
synth-pdb --sequence TRP-TYR-PHE --minimize --solvent explicit --solvent-padding 1.5 --keep-solvent --output full_water_box.pdb
# 🚀 EXTREME Hardware Limit Test
# Generate a large 50-residue sequence, bury it in a massive 2.5 nm water box,
# and run 10,000 steps of Langevin Dynamics equilibration.
# WARNING: This will generate >50,000 atoms and heavily tax your CPU/GPU!
synth-pdb --length 50 --conformation random --minimize --equilibrate --md-steps 10000 --solvent explicit --solvent-padding 2.5 --keep-solvent --output extreme_limit_test.pdbsynth-pdb is designed to be a high-performance "Data Factory" for Training Protein AI models. It can generate thousands of unique, physically plausible protein structures in seconds—bypassing the bottleneck of parsing millions of PDB files from disk.
Using the BatchedGenerator module, the tool uses SIMD/Vectorized math (NeRF algorithm) to build peptide backbones in parallel.
Transition from biological coordinates to Deep Learning tensors instantly. Our BatchedPeptide output is C-Contiguous, allowing tools like PyTorch and JAX to map the memory without copying data.
from synth_pdb.batch_generator import BatchedGenerator
import torch
# Generate 1,000 structures in milliseconds
bg = BatchedGenerator("ALA-GLY-SER-TRP", n_batch=1000)
batch = bg.generate_batch()
# Instant PyTorch Handover (Shared RAM)
coords_tensor = torch.from_numpy(batch.coords).float()- AI Protein Data Factory:
For detailed examples of how to load generated data into your favorite framework without any performance overhead, see our specialized handover notebooks:
- JAX Handover - Zero-copy using
jax.numpy.asarray. - PyTorch Handover - Unified memory mapping with
torch.from_numpy. - MLX Handover - Optimized for Apple Silicon (M-series CPUs/GPUs).
# Refine steric clashes (5 iterations)
synth-pdb --length 30 --refine-clashes 5 --output refined.pdb
# Combined: best of 10 + refinement
synth-pdb --length 25 --best-of-N 10 --refine-clashes 3 --output optimized.pdbGenerate structures that mimic real protein motifs for educational demonstrations:
# Collagen-like triple helix motif (polyproline II)
# Collagen is rich in proline and glycine with PPII conformation
synth-pdb --sequence "GPGPPGPPGPPGPPGPPGPP" --conformation ppii --output collagen_like.pdb
# Silk fibroin-like beta sheet
# Silk proteins contain repeating (GAGAGS) motifs forming beta sheets
synth-pdb --sequence "GAGAGSGAGAGSGAGAGS" --conformation beta --output silk_like.pdb
# Amyloid fibril-like beta structure
# Amyloid fibrils are rich in beta sheets, often with hydrophobic residues
synth-pdb --sequence "LVEALYLVCGERGFFYTPKA" --conformation beta --best-of-N 10 --output amyloid_like.pdb
# Leucine zipper motif (alpha helix)
# Leucine zippers are alpha-helical with leucine repeats every 7 residues
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" --conformation alpha --output leucine_zipper.pdb
# Intrinsically disordered region (random conformation)
# IDRs lack stable structure, rich in charged/polar residues
synth-pdb --sequence "GGSEGGSEGGSEGGSEGGSE" --conformation random --output disordered_region.pdb
# Transmembrane helix-like structure (extended alpha helix)
# Membrane-spanning regions are often long alpha helices with hydrophobic residues
synth-pdb --sequence "LVIVLLVIVLLVIVLLVIVL" --conformation alpha --output transmembrane_like.pdb
# Beta-turn rich structure (mixed conformations)
# Proline and glycine favor turns and loops
synth-pdb --sequence "GPGPGPGPGPGPGPGP" --conformation random --output beta_turn_rich.pdb
# Elastin-like peptide (extended/random)
# Elastin contains repeating VPGVG motifs with flexible structure
synth-pdb --sequence "VPGVGVPGVGVPGVGVPGVG" --conformation extended --output elastin_like.pdb
# Antimicrobial peptide-like (alpha helix)
# Many AMPs are short amphipathic alpha helices
synth-pdb --sequence "KWKLFKKIGAVLKVL" --conformation alpha --validate --output amp_like.pdb
# Zinc finger motif-like (mixed structure)
# Zinc fingers have beta sheets and alpha helices
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --conformation random --best-of-N 5 --output zinc_finger_like.pdbEducational Notes:
- These examples demonstrate sequence-structure relationships
- Real proteins would have more complex tertiary structures and post-translational modifications
- Use these for teaching secondary structure concepts, not for actual molecular modeling
- Combine with
--validateto show how different conformations affect structural quality - Try
--best-of-Nand--refine-clashesto explore quality control strategies
These examples are specifically designed to look great in the 3D viewer with --visualize:
# 🧬 Compact Alpha Helix (BEST for visualization)
# Short, tight helix - perfect for interactive viewing
synth-pdb --length 15 --conformation alpha --visualize
# 🔗 Helix-Turn-Helix DNA-Binding Motif
# Classic protein architecture with two helices and a turn
synth-pdb --sequence "AAAAAAGGGAAAAA" --structure "1-6:alpha,7-9:random,10-14:alpha" --visualize
# 🧬 "Textbook" Stabilized Alpha Helix (Salt Bridges)
# Demonstrates charge pairs (Glu-Lys) stabilizing the backbone (i, i+4)
# Use --minimize to geometry-optimize these ionic interactions
synth-pdb --sequence "EAAKEAAKEAAKEAAK" --conformation alpha --minimize --cap-termini --visualize
# 🔗 Zinc Finger with Metal Coordination
# See the Zinc ion (Zn2+) automatically coordinated by Cys/His residues!
# The --minimize flag applies harmonic constraints to the metal center.
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --structure "1-10:beta,11-20:alpha" --metal-ions auto --minimize --visualize
# 🎀 Refined Beta Hairpin
# Two antiparallel beta strands connected by a turn, relaxed with physics
synth-pdb --sequence "VVVVVGGVVVVV" --structure "1-5:beta,6-8:random,9-12:beta" --minimize --visualize
# 🧪 Polyproline II Helix (Collagen-like)
# Left-handed helix, compact and visually distinct
synth-pdb --sequence "GPGPPGPPGPPGPP" --conformation ppii --minimize --visualize
# 🧪 The "Kitchen Sink" (Features Demo)
# Combines distinct secondary structures (Helix, Sheet) with a Type I Beta Turn and PTMs.
# Look for the magenta helix, purple turn, and orange phosphorylated residues (SEP/TPO/PTR).
synth-pdb --length 25 --structure "1-10:alpha,11-14:typeI,15-25:beta" --phosphorylation-rate 0.3 --visualize
# ⭕ The "Molecular Hoop" (Macrocycle)
# A simple flexible ring of Glycines. Perfect for visualizing ring closure.
synth-pdb --sequence "GGGGGGGGGGGG" --cyclic --minimize --visualizeVisualization Tips:
- Best conformations for viewing:
alpha(most compact),ppii(distinctive shape) - Optimal length: 10-20 residues for clear visualization
- In the viewer: Use "Cartoon" style and "Spectrum" color for best results
- Interactive: Rotate with left-click, zoom with scroll, pan with right-click
The --structure parameter enables creation of realistic protein-like structures with different conformations in different regions:
# Helix-turn-helix DNA-binding motif
# Two alpha helices connected by a flexible turn region, minimized for realism
synth-pdb --length 25 --structure "1-10:alpha,11-15:random,16-25:alpha" --minimize --output helix_turn_helix.pdb
# Beta-alpha-beta fold unit
# Common protein architecture with sheet-helix-sheet
synth-pdb --length 30 --structure "1-10:beta,11-15:random,16-25:alpha,26-30:beta" --minimize --output bab_fold.pdb
# Zinc finger with realistic structure
# Beta sheet + alpha helix (actual zinc finger architecture)
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --structure "1-5:beta,6-10:random,11-20:alpha" --minimize --output zinc_finger_realistic.pdb
# Immunoglobulin domain
# Multiple beta sheets connected by loops (antibody-like)
synth-pdb --length 40 --structure "1-8:beta,9-12:random,13-20:beta,21-24:random,25-32:beta,33-40:random" --minimize --output ig_domain.pdb
# Coiled-coil with flexible linker
# Two helical regions connected by disordered linker
synth-pdb --length 50 --structure "1-20:alpha,21-30:random,31-50:alpha" --minimize --output coiled_coil.pdb
# Intrinsically disordered region with structured domain
# Disordered N-terminus, structured C-terminus (common in signaling proteins)
synth-pdb --length 40 --structure "1-15:random,16-40:alpha" --minimize --output idr_with_domain.pdb
# Collagen-like with flexibility
# PPII helix with occasional flexible regions (more realistic than uniform)
synth-pdb --sequence "GPGPPGPPGPPGPPGPPGPP" --structure "1-6:ppii,7-9:random,10-20:ppii" --output collagen_flexible.pdb
# Beta-hairpin motif
# Two antiparallel beta strands connected by a turn
synth-pdb --length 20 --structure "1-7:beta,8-12:random,13-20:beta" --refine-clashes 5 --output beta_hairpin.pdbWhy This Matters:
- Real proteins have mixed secondary structures, not uniform conformations
- These examples are much more realistic than single-conformation structures
- Useful for teaching protein architecture and domain organization
- Great for testing structure analysis tools with realistic inputs
- Demonstrates how sequence and structure work together
These comprehensive examples demonstrate how to use synth-pdb to model specific biological features found in well-known proteins.
1. Glucagon (Alpha Helix Hormone) 29 residues | PDB: 1GCN Glucagon is a peptide hormone that raises glucose levels. It folds into a characteristic alpha helix.
synth-pdb --sequence HSQGTFTSDYSKYLDSRRAQDFVQWLMNT --conformation alpha --refine-clashes 0 --output glucagon.pdbEducational Concept: Studying alpha-helical packing and amphipathicity.
2. Melittin (Bent Helix / Hinge) 26 residues | PDB: 2MLT The principal toxin in bee venom. It forms two alpha helices separated by a "hinge" region, allowing it to puncture membranes.
synth-pdb --sequence GIGAVLKVLTTGLPALISWIKRKRQQ --structure "1-11:alpha,12-14:random,15-26:alpha" --refine-clashes 50 --output melittin.pdbEducational Concept: Modeling non-linear secondary structures and flexible linkers (hinges).
3. Bovine Pancreatic Trypsin Inhibitor (BPTI) (Disulfide Bonds) 58 residues | PDB: 1BPI A classic model for protein folding studies ("The Hydrogen Atom of Protein Folding"). It is stabilized by three disulfide bonds.
synth-pdb --sequence RPDFCLEPPYTGPCKARIIRYFYNAKAGLCQTFVYGGCRAKRNNFKSAEDCMRTCGGA --conformation random --minimize --visualize --output bpti.pdbEducational Concept: Automatic detection of disulfide bonds (SSBOND records). The --minimize flag brings cysteine sulfurs into proper bonding distance (2.0 Å).
4. Ubiquitin (Complex Mixed Fold) 76 residues | PDB: 1UBQ A highly conserved regulatory protein with a complex mixed alpha/beta fold (beta grasp fold).
synth-pdb --sequence MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG --structure "1-7:beta,12-16:beta,23-34:alpha,41-45:beta,48-49:beta,56-59:alpha,66-70:beta" --minimize --best-of-N 5 --output ubiquitin.pdbEducational Concept: Generating complex, multi-domain topologies. Physics-based minimization (--minimize) resolves steric clashes better than geometric heuristics alone.
5. SFTI-1 (Sunflower Trypsin Inhibitor) 14 residues | PDB: 1SFI A small, potent protease inhibitor that is both cyclic and stabilized by a disulfide bond.
synth-pdb --sequence "GRCTKSIPPICFPD" --cyclic --minimize --visualize --output sfti1.pdbEducational Concept: Combining multiple stabilizing modifications (Cyclization + Disulfide Bonds) to create a rigid, functional scaffold.
6. Gramicidin S (D-Amino Acid Antibiotic)
10 residues | PDB: 1TK2
A powerful cyclic antibiotic produced by soil bacteria. It contains the rare D-Phenylalanine (D-PHE) which is critical for its "beta-sheet-like" hairpins.
synth-pdb --sequence "VAL-ORN-LEU-D-PHE-PRO-VAL-ORN-LEU-D-PHE-PRO" --cyclic --minimize --visualize --output gramicidin_s.pdbNote: This utilizes ORN (Ornithine) if supported, or sub for LYS. The key is the D-PHE residue. Educational Concept: Using D-amino acids to induce specific turns and achieve antimicrobial activity through membrane disruption.
These larger structures demonstrate domain organization and fibrous protein architectures.
1. "Synthetic Spectrin" (Multi-Domain Repeat) ~150 Residues Spectrin is a cytoskeletal protein made of repeating triple-helical bundles. We can simulate a simplified version: three distinct alpha-helical domains connected by flexible linkers.
synth-pdb --length 150 --structure "1-40:alpha,41-50:random,51-90:alpha,91-100:random,101-140:alpha,141-150:random" --minimize --visualize --output synthetic_spectrin.pdbEducational Concept: Demonstrates "beads on a string" domain organization and stable inter-domain flexibility.
2. "Titin Segment" (Poly-Beta Repeat) ~120 Residues Titin acts as a molecular spring in muscle, made of distinct Ig-like (beta sheet) domains.
synth-pdb --length 120 --structure "1-30:beta,31-40:random,41-70:beta,71-80:random,81-110:beta,111-120:random" --minimize --visualize --output titin_segment.pdbEducational Concept: Shows distinct rigid beta-regions separated by disordered "hinges", mimicking force-bearing structural proteins.
3. "Giant Coiled-Coil" (The Molecular Rod) ~100 Residues A super-long continuous alpha helix, modeled after Myosin tails or Tropomyosin.
synth-pdb --sequence "LKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKE" --conformation alpha --minimize --visualize --output long_coil.pdbEducational Concept: A massive, rigid rod where the helical groove is clearly visible. Excellent for demonstrating persistence length.
4. "Synthetic Antibody" (The Ultimate Stress Test) 450 Residues Empirical simulation of a full IgG Heavy Chain: 4 Beta-sandwich domains (VH, CH1, CH2, CH3) connected by linkers.
synth-pdb --length 450 --structure "1-100:beta,101-110:random,111-210:beta,211-230:random,231-330:beta,331-340:random,341-440:beta,441-450:random" --minimize --visualize --output synthetic_antibody.pdbNote: This is a computationally intensive task! Energy minimization for ~7000 atoms may take several minutes. Educational Concept: Simulating multi-domain packing and the flexibility of the hinge region (residues 211-230).
# All natural amino acids with validation report
synth-pdb --sequence "ACDEFGHIKLMNPQRSTVWY" --validate --log-level DEBUG
# Test structure for MD simulation pipeline
synth-pdb --length 50 --guarantee-valid --max-attempts 500 --output test_md.pdb
# Benchmark structure with known violations (good for testing validators)
synth-pdb --length 100 --validate --output benchmark.pdbCombine all features to simulate a complete NMR structure determination workflow:
- Generate a sequence.
- Fold it (alpha helix).
- Refine geometry (minimization).
- Simulate experimental data (NOEs and Relaxation).
- Visualize the result.
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" \
--conformation alpha \
--minimize \
--gen-nef --noe-cutoff 6.0 \
--gen-relax --field 800 \
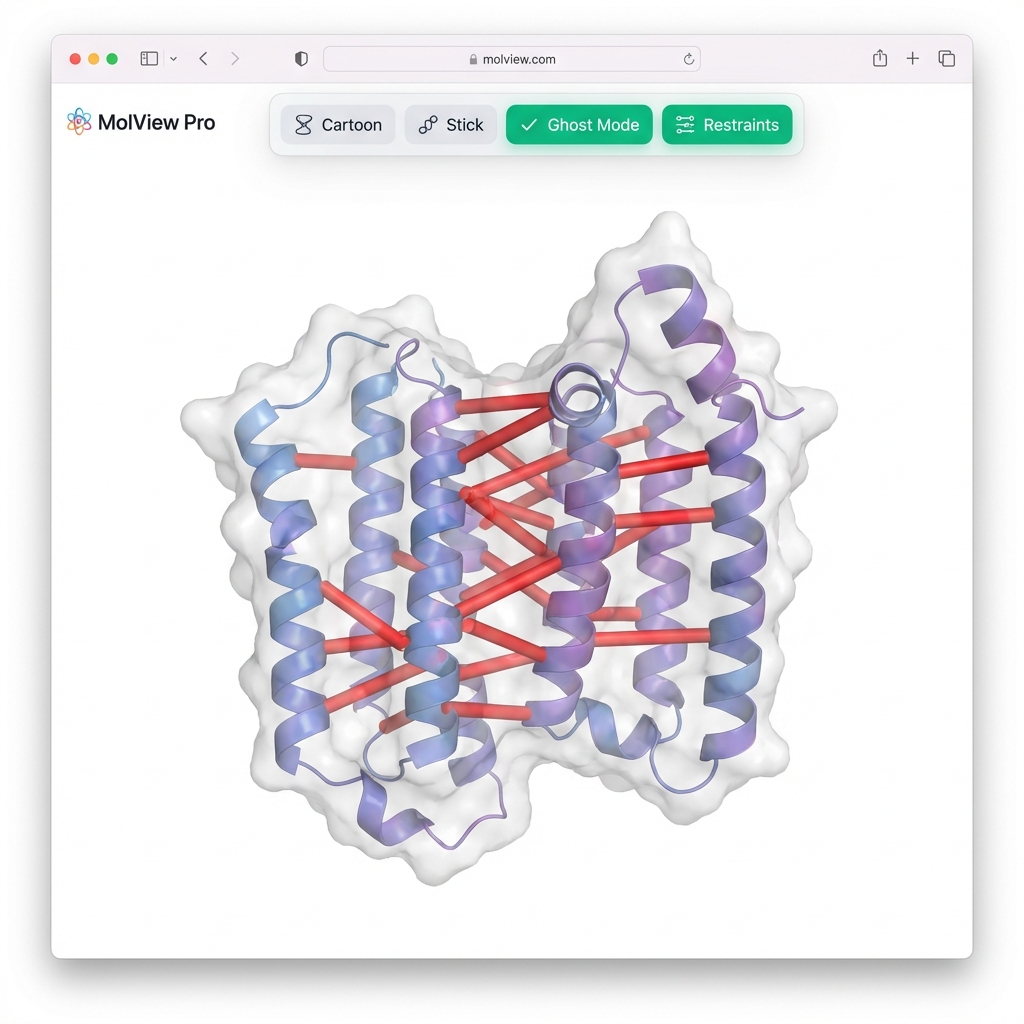
--visualize👀 Viewer Tip: Since you used
--gen-nef, the synthetic NOE restraints will automatically appear as red cylinders connecting the protons. Use the "🔴 Restraints" button in the viewer to toggle them on/off!
A classic biophysical motif where one face of the helix is hydrophobic (L, V, I) and the other is hydrophilic (K, E, R).
# Generate and Minimize
synth-pdb --sequence "LKWLKRLLKWLKRLLKWLKRL" --conformation alpha --minimize --visualizeIn the viewer: Switch to "Sphere" style and "Element" color. You will see the "greasy" hydrophobic patch (Carbon-rich) clearly separated from the charged residues (Nitrogen/Oxygen-rich). This "hydrophobic moment" drives membrane binding!
🎓 Academic Note - "Amphipathic": From Greek amphi (both) and pathos (feeling). An amphipathic helix has a "split personality":
- Hydrophobic Face (L, V, I, F): Hates water. Buries itself inside the protein core or membrane.
- Hydrophilic Face (K, R, E, D): Loves water. Faces the solvent to keep the protein soluble. This duality is the fundamental force driving protein folding! 🧬🌗
When --validate is enabled, the tool checks for:
-
Bond Lengths: Compares N-CA, CA-C, C-N, C-O distances against standard values (±0.05 Å tolerance)
-
Bond Angles: Validates N-CA-C, CA-C-N, CA-C-O angles (±5° tolerance)
-
Ramachandran Angles: Checks phi/psi dihedral angles against MolProbity-defined polygonal regions
- Categories: General, Glycine, Proline, Pre-Proline
- Levels: Distinguishes between Favored, Allowed, and Outlier status
-
Steric Clashes: Detects atoms that are too close
- Minimum distance rule: ≥2.0 Å between any atoms
- van der Waals overlap: atoms closer than sum of vdW radii
-
Peptide Plane Planarity: Checks omega (ω) dihedral angles
- Trans: ~180° (±30° tolerance)
- Cis: ~0° (±30° tolerance)
-
Sequence Improbabilities: Flags unusual sequence patterns
- Charge clusters (4+ consecutive charged residues)
- Long hydrophobic stretches (8+ residues)
- Odd cysteine counts (unpaired cysteines)
- Poly-proline or poly-glycine runs
-
Chirality: Validates L-amino acid stereochemistry
- Checks improper dihedral angle N-CA-C-CB
- L-amino acids should have proper chirality (improper dihedral ±60° to ±120°)
- Glycine is automatically exempt (no CB atom)
- Detects incorrect stereochemistry (D-amino acids)
The --refine-clashes option uses an iterative approach:
- Identifies clashing atom pairs
- Slightly adjusts positions to increase separation
- Re-validates structure
- Stops when no improvement or max iterations reached
Note: Refinement focuses on steric clashes and may introduce other violations. Use in combination with
--best-of-Nfor better results.
- Full Atomic Model: All backbone atoms (N, CA, C, O) + side-chain heavy atoms + hydrogens
- Geometry: Linear alpha-helix conformation along the X-axis
- Chain ID: Always 'A'
- Residue Numbering: Sequential from 1
- Terminal Modifications: N-terminal and C-terminal hydrogens/oxygens included
Each atom line follows the standard PDB format. The B-factor (Temperature Factor) is stored in columns 61-66.
ATOM 1 N ALA A 1 -2.193 1.858 1.271 0.85 56.71 N
ATOM 5 CB ALA A 1 0.241 1.845 1.013 0.85 86.14 C
^^^^ ^^^^^
Occpy B-Fact
-
Occupancy (0.85): Reflects the Order Parameter (
$S^2$ ) if calculated, or default. - B-Factor (56.71 vs 86.14): Reflects atomic mobility. Note how the side-chain atom (CB) has a higher B-factor than the backbone (N), indicating greater flexibility.
Generated PDB files include standard header records:
HEADER PEPTIDE <DATE>
TITLE GENERATED LINEAR PEPTIDE OF LENGTH <N>
REMARK 1 This PDB file was generated by the CLI 'synth-pdb' tool.
REMARK 2 It represents a simplified model of a linear peptide chain.
REMARK 2 Coordinates are idealized and do not reflect real-world physics.
REMARK 3 GENERATION PARAMETERS:
REMARK 3 Command: synth-pdb --length 10 --validate ...
The REMARK 3 records store the exact command-line arguments used for reproducibility.
When --validate is used, violations are reported:
WARNING --- PDB Validation Report for /path/to/file.pdb ---
WARNING Final PDB has 5 violations.
WARNING Bond length violation: N-1-A to CA-1-A. Distance: 1.52Å, Expected: 1.46ű0.05Å
WARNING Steric clash (min distance): Atoms CA-3-A and CB-3-A are too close (1.85Å)...
✅ Appropriate Uses:
- Testing PDB parsers and file I/O
- Benchmarking structure validation tools
- Educational demonstrations of protein structure concepts
- Generating test datasets for bioinformatics pipelines
- Placeholder structures for software development
❌ Inappropriate Uses:
- Homology modeling templates
- Drug docking studies
- Experimental predictions
- Publication-quality structures
Real protein structures require sophisticated methods like:
- Molecular dynamics with force fields (AMBER, CHARMM)
- Quantum mechanics calculations (DFT)
- Energy minimization and conformational search
- Crystallographic or NMR experimental data
-
Topology:
- Primarily generates linear variations or simple disulfide-bonded loops.
- Does not perform de novo folding (prediction of tertiary structure from sequence).
- Multi-chain complexes are currently limited to simple docking preparations.
-
Geometry:
- Default Mode: Uses idealized internal coordinates (perfect bond lengths/angles).
- Physically Realistic Mode (
--minimize): Resolves this by relaxing the structure with OpenMM, but is computationally more expensive.
-
Rotamer Library:
- Backbone-Dependent: Fully implemented for All 20 Amino Acids.
- Mechanism: Checks local secondary structure (Alpha/Beta) to select rotamers that avoid backbone clashes.
- Rare Rotamers: Very rare side-chain conformations (<1% probability) may be undersampled.
-
Environmental Effects:
- Solvent: Uses Implicit Solvent (OBC2) to model water screening, but lacks explicit water molecules.
- Membranes: No lipid bilayer simulation for transmembrane proteins.
- Ramachandran Regions: Uses simplified rectangular boundaries for valid phi/psi regions. While faster, this is less rigorous than the contoured probability density functions used by MolProbity.
- Electrostatics: Basic clash detection does not account for long-range electrostatic repulsion/attraction (though
--minimizedoes). - Protonation: Simple pH-based titration (His/Asp/Glu) without full pKa calculation.
There is an important distinction between the "Decoys" generated by this tool and a traditional "NMR Ensemble":
- NMR Ensemble: A set of structures (usually 20) that all satisfy experimental restraints (NOEs) and have converged to the same fold. They represent the precision of the structure determination.
- Decoys (Conformational Ensemble): A set of independent structures generated to sample the conformational space. They often have high RMSD (diversity) and represent the search space.
synth-pdb --mode decoys generates the latter: independent snapshots. To create a pseudo-NMR ensemble, use --rmsd-max 2.0 to filter for similar structures.
-
--guarantee-validmay never converge for long sequences (>50 residues)- Combinatorial explosion of possible violations
- Consider using
--best-of-Ninstead
-
--refine-clashesis iterative and may be slow for large structures- Each iteration requires full re-validation
-
Validation runtime scales with sequence length (O(N²) for steric clashes)
# All tests
pytest -v
# With coverage
pytest --cov=synth_pdb --cov-report=term-missing
# Specific test file
pytest tests/test_generator.py -vTest Coverage: 95% overall
- 729 tests covering generation, validation, CLI, and edge cases
synth-pdb/
├── synth_pdb/
│ ├── __init__.py
│ ├── main.py # CLI entry point
│ ├── generator.py # PDB structure generation
│ ├── validator.py # Validation checks
│ └── data.py # Constants and rotamer library
├── tests/
│ ├── test_generator.py
│ ├── test_generator_rotamer.py
│ ├── test_validator.py
│ └── test_main_cli.py
├── setup.py
└── README.md
For students and researchers interested in the physics behind the code, here are key seminal papers:
-
Cis-Proline (~5% Frequency):
- MacArthur, M. W., & Thornton, J. M. (1991). Influence of proline residues on protein conformation. J Mol Biol, 218(2), 397-412.
- Weiss, M. S., et al. (1998). Cis-proline. Acta Cryst D, 54, 323-329.
-
Macrocyclization & Cyclic Peptides:
- Horton, D. A., et al. (2003). The combinatorial synthesis of bicyclic peptides. Chem. Rev., 103(3), 893-930. (Seminal review on macrocycles).
- Craik, D. J., et al. (2013). The future of peptide-based drugs. Chem. Biol. Drug Des., 81(1), 136-147.
-
NMR Structure Validation & Chirality:
- Montelione, G. T., et al. (2013). Recommendations of the wwPDB NMR Validation Task Force. Structure, 21(9), 1563-1570. (Defines standards for geometric validation).
- Bhattacharya, A., & Montelione, G. T. (2011). PDBStat: a server for validation of protein NMR structures.
-
Nuclear Overhauser Effect (NOE) &
$r^{-6}$ :- Wüthrich, K. (1986). NMR of Proteins and Nucleic Acids. Wiley-Interscience. (The definitive text).
- Wüthrich, K. (2003). Nobel Lecture: NMR Studies of Protein Structure and Dynamics.
-
Chemical Shift Prediction (SPARTA) & Referencing (DSS):
- Shen, Y., & Bax, A. (2010). SPARTA+: a modest improvement in empirical NMR chemical shift prediction... J Biomol NMR, 48, 13-22.
- Markley, J. L., et al. (1998). Recommendations for the presentation of NMR structures... (IUPAC). Pure Appl Chem, 70(1), 117-142. (Defined DSS as the standard).
-
Internal Dynamics & Model-Free Formalism:
- Lipari, G., & Szabo, A. (1982). Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc., 104(17), 4546–4559. (The foundational theory).
- Kay, L. E., Torchia, D. A., & Bax, A. (1989). Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy... Biochemistry, 28(23), 8972-8979. (The seminal application to proteins).
- Dunbrack, R. L., & Cohen, F. E. (1997). Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Science, 6(8), 1661–1681.
- Used for: Rotamer libraries and side-chain probability distributions.
- Parsons, J., et al. (2005). Practical conversion from torsion space to Cartesian space for in silico protein synthesis. Journal of Computational Chemistry, 26(10), 1063–1068.
- Used for: The NeRF (Natural Extension Reference Frame) algorithm for backbone construction.
- MacArthur, M. W., & Thornton, J. M. (1991). Influence of proline residues on protein conformation. Journal of Molecular Biology, 218(2), 397-412.
- Used for: Cis-Proline isomerization statistics (~5% cis frequency).
- Homeyer, N., et al. (2006). AMBER force-field parameters for phosphorylated amino acids... Journal of Molecular Modeling, 12(3), 281-289.
- Used for: PTM physics parameters (SEP, TPO, PTR) in OpenMM.
- Smith, D. M. (2001). Protein Composition and Structure. Encyclopedia of Life Sciences.
- Used for: Biological amino acid frequency data.
-
Lipari, G., & Szabo, A. (1982). Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. Journal of the American Chemical Society, 104(17), 4546–4559.
- Used for: Calculating
$S^2$ order parameters and relaxation rates ($R_1, R_2, NOE$ ).
- Used for: Calculating
-
Wishart, D. S., et al. (1995). 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. Journal of Biomolecular NMR, 6, 135–140.
- Used for: Random coil chemical shift baselines.
-
Cavanagh, J., et al. (2007). Protein NMR Spectroscopy: Principles and Practice. Academic Press.
- Used for: General NMR theory and relaxation equations.
- Williams, C. J., et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Science, 27(1), 293–315.
- Used for: Ramachandran polygon definitions and validation criteria.
- Lovell, S. C., et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins: Structure, Function and Bioinformatics, 50(3), 437–450.
- Used for: Early reference for Ramachandran validation concepts.
This section provides definitions and seminal references for the biophysical and computational terms used throughout synth-pdb.
| Term | Definition | Reference |
|---|---|---|
| PDB | Protein Data Bank. The global repository for 3D structural data of proteins, nucleic acids, and complex assemblies. | Berman, H. M., et al. (2000). Nucleic Acids Res. |
| CASP | Critical Assessment of Structure Prediction. A community-wide experiment held every two years to establish the state-of-the-art in protein structure modeling. | Kryshtafovych, A., et al. (2021). Proteins. |
| NOE | Nuclear Overhauser Effect. A phenomenon where magnetization is transferred between spins through space, allowing for the measure of inter-atomic distances (r⁻⁶ dependency). | Wüthrich, K. (1986). NMR of Proteins and Nucleic Acids. |
| CSI | Chemical Shift Index. A standard method used to deduce protein secondary structure (alpha helix vs. beta sheet) from detected NMR chemical shift deviations. | Wishart, D. S., et al. (1992). Biochemistry. |
| NEF | NMR Exchange Format. A unified, open standard for the exchange of NMR restraint data among various software packages. | Gutmanas, A., et al. (2015). Nat. Struct. Mol. Biol. |
| NeRF | Natural Extension Reference Frame. An algorithm for rapidly constructing 3D Cartesian coordinates from internal coordinates (bond lengths, angles, and dihedrals). | Parsons, J., et al. (2005). J. Comput. Chem. |
| S² | Model-Free Order Parameter. A value between 0 (random) and 1 (rigid) that describes the degree of spatial restriction of local backbone motion. | Lipari, G., & Szabo, A. (1982). J. Am. Chem. Soc. |
| OBC2 | Onufriev-Bashford-Case. A computationally efficient implicit solvent model (Generalized Born) used to simulate the screening effect of water. | Onufriev, A., et al. (2004). Proteins. |
| AMBER | Assisted Model Building with Energy Refinement. A widely-used suite of molecular simulation programs and force fields for biomolecules. | Case, D. A., et al. (2005). J. Comput. Chem. |
| Macrocycle | A cyclic macromolecule or a macromolecular network, such as a cyclic peptide or a crown ether. | IUPAC Gold Book. |
This project is provided as-is for educational and testing purposes.
If you use this software in your research, please cite:
@software{synth_pdb,
author = {Elkins, George},
title = {synth-pdb: Realistic Protein Structure Generator},
year = {2026},
url = {https://github.com/elkins/synth-pdb}
}This project relies on the following open-source scientific software:
- OpenMM: High-performance molecular dynamics toolkit used for physics-based energy minimization (Implicit Solvent/OBC2).
- Biotite: Comprehensive library for structural biology involved in PDB IO, atom manipulation, and geometric analysis.
- 3Dmol.js: JavaScript library for molecular visualization used in the
--visualizebrowser-based viewer. - NumPy: Fundamental package for scientific computing and matrix operations.
These external tools can import the data generated by synth-pdb:
- CCPNMR Analysis: Premier software for NMR data analysis, assignment, and structure calculation (Native NEF support).
- CYANA: Automated NMR structure calculation.
- XPLOR-NIH: Biomolecular structure determination.
-
Protein Structure Validation Suite (PSVS)
- Bhattacharya, A., Tejero, R., & Montelione, G. T. (2007). "Evaluating protein structures determined by structural genomics consortia." Proteins: Structure, Function, and Bioinformatics, 66(4), 778-795.
- Link to Publisher
-
RPF Scores (Recall, Precision, F-measure)
- Huang, Y. J., Powers, R., & Montelione, G. T. (2005). "Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics." Journal of the American Chemical Society, 127(6), 1665-1674.
- Link to Publisher
-
DP Score (Discriminant Power)
- Huang, Y. J., Tejero, R., Powers, R., & Montelione, G. T. (2006). "A topology-constrained distance network algorithm for protein structure determination from NOESY data." Proteins: Structure, Function, and Bioinformatics, 62(3), 587-603.
- Link to Publisher
- NMR Exchange Format (NEF)
- Gutmanas, A., et al. (2015). "NMR Exchange Format: a unified and open standard for representation of NMR restraint data." Nature Structural & Molecular Biology, 22, 433–434.
- Link to Publisher
- Extension Proposal: "Proposal For Incorporating NMR Relaxation Data In NEF" (GitHub PDF)
- Ramachandran Plot: Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). "Stereochemistry of polypeptide chain configurations." Journal of Molecular Biology.
- Rotamer Libraries: Dunbrack, R. L. (2002). "Rotamer libraries in the 21st century." Current Opinion in Structural Biology.
- PDB Format Specification: wwPDB Documentation
- IUPAC Nomenclature: IUPAC-IUBMB Joint Commission on Biochemical Nomenclature
This section explains the science behind the --minimize feature.
Imagine a ball rolling on a hilly landscape.
- Height = Potential Energy (unstable)
- Valleys = Stable conformations (low energy)
- Gravity = Interactions between atoms (forces)
Energy Minimization is the process of moving atoms "downhill" to find the nearest stable shape.
High Energy
(Unstable)
|
/ \ Forces push atoms "downhill"
/ \ (Gradient Descent)
/ \
/ \___
/ \
/ \__ Low Energy
/ (Stable / Minimized)
Proteins exist in water. Simulating every water molecule is expensive.
- Explicit Solvent: Simulating thousands of H2O molecules. (Accurate but Slow)
- Implicit Solvent: Treating water as a mathematical continuous field that shields charges. (Fast and Good Approximation)
synth-pdb uses Implicit Solvent (OBC2) to get realistic results quickly.
How synth-pdb builds a protein from scratch:
[User] -> [Generator] -> [Geometry Builder] -> [Sidechain Packer] -> [Energy Minimizer] -> [PDB File]
^ | | |
| (N-CA-C-O) (Rotamers) (OpenMM)
| | |
+---------------------------------------+----------------------+
The "Order Parameter" (
-
$S^2 = 1.0$ : Perfectly rigid (Low B-factor). -
$S^2 < 0.8$ : Flexible (High B-factor). Use this to validate Model-Free analysis software.
Generates predicted chemical shifts (
We implement a simplified version of the SPARTA algorithm:
- Base Value: Random Coil shift (Wishart et al.).
-
Offset: Based on the exact
$\phi, \psi$ angles of the generated structure.-
Helix:
$C_\alpha$ +3.1 ppm,$N$ -1.5 ppm. -
Sheet:
$C_\alpha$ -1.5 ppm,$N$ +1.2 ppm.
-
Helix:
This allows you to verify that your assignment software (e.g. CCPNMR) correctly identifies secondary structure from shifts ("CSI" method).
The output _shifts.nef file mimics a deposited BMRB entry, enabling direct import into:
- CCPNMR Analysis V3 (Drag and drop!)
- CYANA (for automated assignment)
- CS-Rosetta (for structure calculation)
--gen-shifts: Enable chemical shift prediction.--shift-output <filename>: Optional. Specify a custom output filename (default:[name]_shifts.nef).
Example:
synth-pdb --sequence "ACDEF" --gen-shifts --shift-output custom_shifts.nefGenerates scalar couplings (
Primarily predicts the HN-HA coupling, which is a powerful indicator of secondary structure:
- Alpha Helix: Small values (< 6 Hz)
- Beta Sheet: Large values (> 8 Hz)
Outputs: A CSV file (_couplings.csv) containing res_id, residue, and predicted J_HN_HA values.
If you are coming from an NMR background (XPLOR-NIH, CYANA, CNS):
-
Structure Calculation vs. Generation:
synth-pdbmimics the final stage of an NMR structure calculation: Geometry Regularization (minimization in implicit solvent). - Proton Detection: Unlike X-ray, NMR relies on 1H spins. That's why we explicitly add hydrogens before minimization—they are the "eyes" of the forcefield, just as they are for NOEs.
-
Ensembles: Use
--mode decoysto generate an ensemble of structures. This is analogous to the "bundle" of low-energy structures you calculate to satisfy NOE restraints. -
Order Parameters: The generated B-factors in
synth-pdbfollow the same pattern as Order Parameters ($S^2$ ) or atomic RMSD across an ensemble (low in core, high in tails).
For production-quality structure generation, consider:
- MODELLER (homology modeling)
- Rosetta (de novo structure prediction)
- AlphaFold (AI-based prediction)
- PyMOL/Chimera (structure visualization and manipulation)
- B-factor: Temperature factor (\pi^2\langle u^2 \rangle$) measuring atomic displacement/flexibility.
-
Backbone-Dependent Rotamer: A side-chain conformation probability that depends on the local backbone angles (
$\phi, \psi$ ). -
Chi Angles (
$\chi$ ): Dihedral angles describing side-chain conformation. - MolProbity: A structure validation web service providing the gold standard for Ramachandran and rotamer analysis.
- OBC2: Onufriev-Bashford-Case model 2. An implicit solvent model used in OpenMM.
-
Phi/Psi (
$\phi, \psi$ ): Backbone dihedral angles.$\phi$ (N-C$\alpha$),$\psi$ (C$\alpha569XilsC). -
Ramachandran Plot: A 2D plot of
$\phi$ vs$\psi$ angles showing allowed backbone conformations. - Pre-Proline: The residue preceding a Proline. It has restricted conformational freedom due to steric clash with the Proline ring.
- Rotamer: Short for "Rotational Isomer". Preferred, low-energy side-chain conformations.
- SASA: Solvent Accessible Surface Area. The surface area of a biomolecule that is accessible to a solvent (like water). Used to determine if a residue is buried (low SASA) or exposed (high SASA).
- Top8000: A high-quality dataset of ~8000 protein chains (resolution < 2.0Å, low homology) used to derive accurate Ramachandran contours and rotamer libraries. (Reference: Lovell et al., 2003).
This project's validation logic is inspired by the foundational work of the Richardson Lab (Duke University) and the Dunbrack Lab (Fox Chase Cancer Center).
-
Ramachandran Validation (Top8000):
Lovell, S. C., et al. (2003). "Structure validation by Calpha geometry: phi,psi and Cbeta deviation." Proteins: Structure, Function, and Bioinformatics, 50(3), 437–450.Used for:
PDBValidatorRamachandran polygons. -
MolProbity:
Chen, V. B., et al. (2010). "MolProbity: all-atom structure validation for macromolecular crystallography." Acta Crystallographica Section D, 66(1), 12–21.Used for: Validation philosophy (favored/allowed/outlier classification).
-
Backbone-Dependent Rotamers:
Dunbrack, R. L., & Cohen, F. E. (1997). "Bayesian statistical analysis of protein side-chain rotamer preferences." Protein Science, 6(8), 1661–1681.Used for:
synth_pdb.data.BACKBONE_DEPENDENT_ROTAMER_LIBRARY.