Accelerating ultra-large-library docking with deep learning for NirA-targeted drug-molecule discovery.
This repository implements a deep learning-guided virtual screening protocol to identify potential inhibitors of the NirA protein—a mission-critical enzyme for therapeutic targeting. By leveraging an ultra-large chemical library and neural network filtering, this workflow achieves a massive reduction in computational overhead while maintaining high precision.
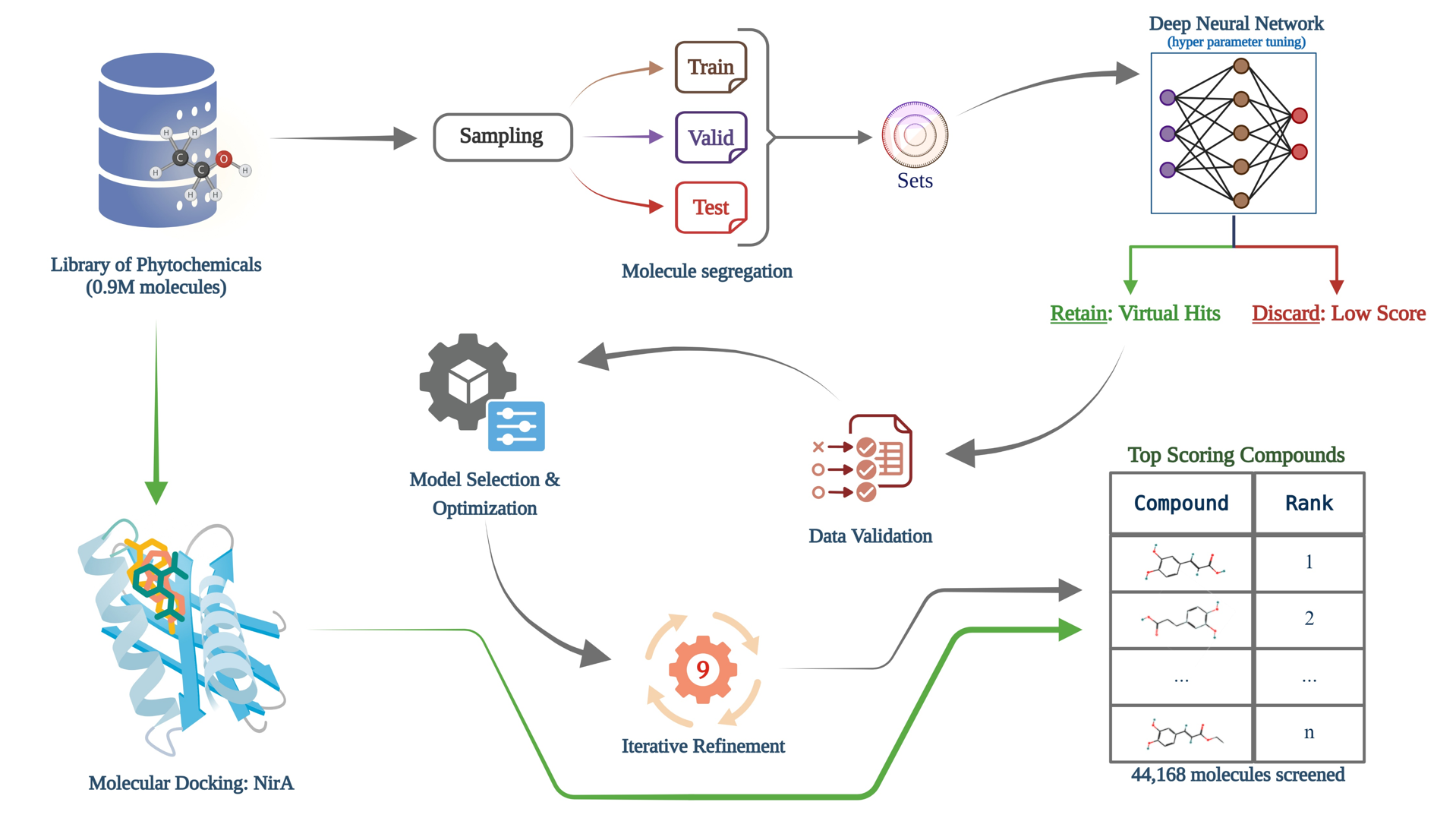
The methodology combines deep neural network-based filtering with molecular docking to rapidly prioritize promising binders from an initial library of ~9 million compounds.
- Sampling from Library: High-diversity sampling from ZINC, DUDE, IMPPAT, and COCONUT.
- Data Preparation: Automated preparation of decoys and actives based on precise molecular weight and activity criteria.
- Dataset Split: Stratified partitioning of ~9 million phytochemicals into Training, Validation, and Testing sets.
- Neural Network Training: Implementation of a feed-forward DNN with optimized hyperparameters for score prediction.
- Virtual Screening: Rapid prediction of docking scores to discard low-affinity compounds and retain high-confidence virtual hits.
- Validation & Refinement: Top hits undergo rigorous validation via molecular docking followed by iterative re-scoring and training rounds.
Breif about the modal :---
Initial Library Size: ~9,000,000 Compounds Virtual Hits Identified: 44,168 Compounds
Model Architecture: Feed-forward Deep Neural Network Optimization: Iterative Refinement & Hyperparameter Tuning
If you utilize this protocol or build upon this research, please cite the original framework and our specialized implementation:
- Original Framework: Gentile, F. et al. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 17, 672–697
- Source Implementation: Original Deep Docking GitHub.
CONTACT for COLLABORATION
thedrsoham[at]gmail[dot]com
"Not the end, but the beginning of a mission."